
Regulation (EU) 2024/1860 Of the European Parliament and of the Council of 13 June 2024 amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards a gradual roll-out of Eudamed, the obligation to inform in case of interruption or discontinuation of supply, and transitional provisions for certain in vitro diagnostic medical devices was published in the Official Journal of the European Union (OJEU) on 9th July 2024. This much anticipated publication is relevant for all medical device and IVD manufacturers marketing, or intending to market, their devices in the EU.
If you’re wondering how to interpret this publication, or how the Eudamed timeline affects your organisation, we’ve got the answers.
Let’s break it down.
Extended Transition to IVDR for Certain IVDs
As long as your IVD device meets the following criteria, you will benefit from the extended transition period outlined in the regulation:
- continued compliance with the IVD Directive 98/79/EC;
- compliance with the IVDR provisions related to PMS and vigilance;
- no significant changes can be made;
- implementation of an IVDR compliant Quality Management System by 26th May 2025;
- a formal conformity assessment application is made with a notified body before:
- 26 May 2025 (class D)
- 26 May 2026 (class C)
- 26 May 2027 (class B and class A sterile)
- a written agreement is in place between the manufacturer and notified body before:
- 26 September 2025 (class D),
- 26 September 2026 (class C) or
- 26 September 2027 (class B and class A sterile).
Compliance with the above provisions entitles IVD manufacturers to the following extended transition periods:
- 31st December 2027 for class D devices;
- 31st December 2028 for class C devices;
- 31st December 2029 for class B
- 31st December 2029 for sterile class A devices.
Points to Note:
- These extensions apply only to legacy devices that have previously been CE marked under the IVD Directive. All new devices must comply with IVDR prior to being placed on the market.
- Non-sterile class A devices do not benefit from any extension. Devices falling into this category already require CE marking under IVDR in order to be placed on the EU market.
Obligations in case of interruption or discontinuation of supply of certain devices – Article 10a
Under the provisions of Regulation (EU) 2024/1860, a new article has been introduced outlining the obligations of manufacturers in the case of an interruption or a discontinuation of the supply of certain devices. This article, a sub-article of Article 10 of both MDR and IVDR, “General obligations of manufacturers”, introduces the requirement for manufacturers to inform their competent authority, or that of its authorised representative, as appropriate, their economic operators, health institutions and healthcare professionals of any anticipated disruption to device availability.
Any anticipated supply chain interruption must be reported at least 6 months in advance of the disruption, other than in exceptional circumstances. The nature of “exceptional circumstances” is not defined in the regulation but should be understood to include unforeseen or unpredictable events. Any reporting delays should be justified and recorded in the manufacturer’s Quality Management System and be made available to competent authorities.
Article 10(a) will apply from 10th January 2025.
The European Database on Medical Devices (EUDAMED) and the Eudamed Timeline
All parties operating under the provisions of the MDR and IVDR will have some level of responsibility in relation to use of Eudamed. Eudamed’s functionality and implementation timelines have been somewhat unclear, following delays in the rollout of the different modules. The original provisions mandated the use of the database only when all modules were declared fully functional. However, following publication of Regulation (EU) 2024/1860, the use of the modules that are already available will become mandatory without the requirement to wait for the readiness and implementation of all modules.
The current status is as follows:
- Actor registration – available since December 2020
- UDI and device registration – available since October 2021
- Notified bodies and certificates – available since October 2021 (with the exception of the functionalities related to scrutiny and the clinical evaluation consultation procedure (CECP)
- Post-market surveillance and vigilance – under development
- Market surveillance – under development
- Clinical investigations and performance studies – under development
On 10th July 2024, one day after the publication of Regulation (EU) 2024/1860, the European Commission released an updated timeline illustrating the current planning for the gradual roll out of Eudamed. The updated Eudamed timeline can be viewed here.
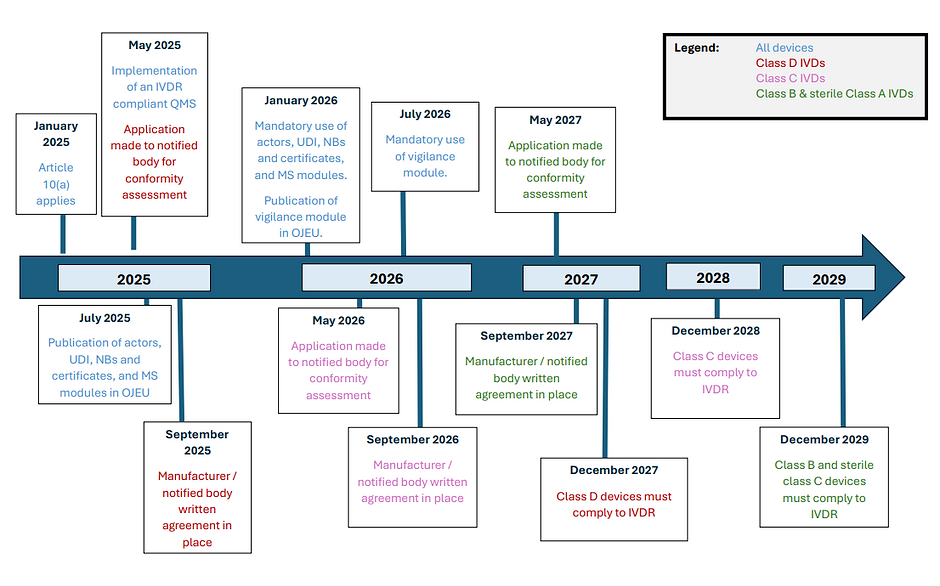
According to the updated timeline, in July 2025, notices are planned for publication in the OJEU for the modules related to actors, UDI/devices, notified bodies and certificates, and market surveillance. Mandatory use of these modules will come into place 6 months later, in January 2026. Also in January 2026, it is planned that the notification regarding the vigilance module will be published in the OJEU. The use of this module will become mandatory in July 2026, following the 6 month transition period.
Final thoughts
The implementation of Regulation (EU) 2024/1860 and the updated Eudamed timeline bring significant changes for medical device and IVD manufacturers. Understanding and complying with these new requirements is essential to ensure uninterrupted market access and regulatory compliance.
The gradual roll-out of Eudamed’s functionalities requires careful planning and timely action. At Trinzo, we are committed to supporting you through these transitions. Contact us today to discuss how we can assist you in navigating these regulatory changes and achieving compliance with confidence.