
Implementation of an effective Post Market Surveillance (PMS) system in medical devices is essential for demonstrating compliance to the EU MDR Quality Management System (QMS) requirements. A recent study (Kearney & McDermott, 2023) reports that the majority of manufacturers of legacy devices intend to use the PMS system as the primary data source for generating sufficient clinical evidence in support of CE marking under the MDR. This finding highlights the importance of implementing and maintaining an effective PMS system.
What is an effective medical device PMS System?
To answer this question, lets first look at the purpose of a PMS system. The purpose of a PMS system is to gather data on real world use of the medical device, to demonstrate that the device continues to be safe and performs as intended. The data must be reliable and of sufficient quality and quantity so that the manufacturer can generate reliable conclusions regarding continuing device quality, safe and performance.
With that in mind, we can say that an effective medical device PMS system facilitates adoption of a total product lifecycle approach whereby the manufacturer continues to monitor the marketed device throughout its lifetime and is prompted to take appropriate actions to ensure quality, safety and performance. An effective PMS system also drives device improvements and can help to identify new uses for the medical device, including opportunities to expand its intended use.

Want to understand how to avoid ticking the boxes, and make PMS really work for you?
If you’re eager to go beyond compliance and unlock the true potential of PMS, join us at our new PMS training course!
Download BrochureWhat are the elements of a PMS System?
The PMS system is comprised of a series of inter-related processes which are designed to gather reactive (passive) and proactive (active) data and information on use of the device. Reactive data sources include data derived from previous Periodic Safety Update Reports (PSURs), Vigilance data including serious incidents and field safety corrective actions, Complaints and unsolicited feedback, Trending data on non-serious incidents and undesirable side effects.
Don’t forget, if you identify a statistically significant increase in non-serious incidents and/or undesirable side effects, you need to submit a trend report to the applicable national competent authority per MEDDEV 2.12-1 (European Commission , 2013).
Proactive data sources are commonly referred to as Post Market Clinical Follow-Up (PMCF) activities and include reviewing regulatory databases and scientific literature to gather data and information on the marketed device and/or similar devices. Other activities include conducting surveys or seeking feedback from health care professionals and patients for the purpose of answering a specific question. Whilst these activities may provide useful and information data, the results must be analysed with caution as they can be subject to bias. PMCF studies are similar to clinical trials, however they occur in the post CE marking phase. For example, the subjects enrolled in the initial pre-marking clinical trial for a new hip implant, continue to be followed for the duration of the implants lifetime. Interestingly, a recent study found that approx. 65% of manufacturers of high risk legacy medical devices intend to conduct PMCF studies in support of MDR. This finding suggests that the clinical data used for initial CE marking under the MDD is not sufficient to demonstrate compliance to the MDR. CE marking (Kearney & McDermott, 2023).
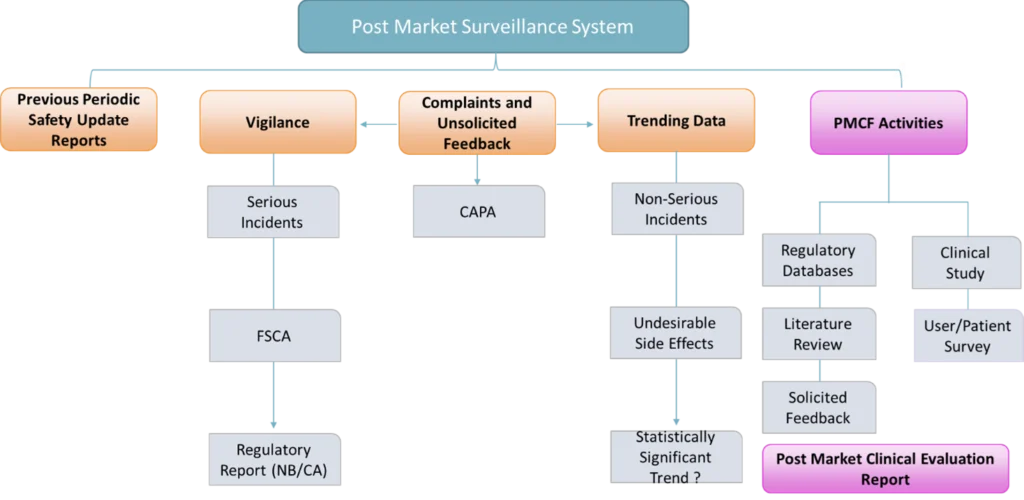
Figure 1: Graphical representation of the elements of a MDR compliant PMS System
How do I implement an effective medical device PMS System?
The PMS system described by MDR 2017/745 aligns with the Plan-Do-Check-Act (PDCA) model (Figure 2).
Plan: Develop a PMS plan which includes all elements defined in Annex III. The plan should clearly describe what data will be collected, how will it be collected and analysed, when will it be done, who is responsible, how will the results and conclusions be presented and what are the communication channels. For additional guidance, refer to ISO/TR 20416:2020 Medical devices — Post-market surveillance for manufacturers. MDCG 2020-7 also provides a useful template for generating a PMCF plan (Medical Device Coordination Group , 2020).
Do: Proactive and reactive data is gathered and analysed using the methodology described in the plan.
Check: Evaluate the results of the data analysis and generate the Periodic Safety Update Report for Class IIa, IIb, III devices per MDR Article 86 and MDCG 2022-21 (Medical Device Coordination Group, 2022), PMS report for Class I devices per MDR Article 85 and/or the PMCF Evaluation Report per MDR Annex XIV, Part B, as applicable. MDCG 2020-8 provides a useful template for the PMCF Evaluation report (Medical Device Coordination Group , 2020).
As part of the check phase, review the adequacy and continued suitability of the PMS plan – do you need to update it?

Act: Depending on the results, the manufacturer may need to initiate a CAPA or FSCA, update risk management file (for example, if there is change in occurrence or a new risks identified etc), update clinical evaluation or might identify opportunities to improve the current design, expand intended use or develop a new device. For further information refer to MDR Article 83 (3).
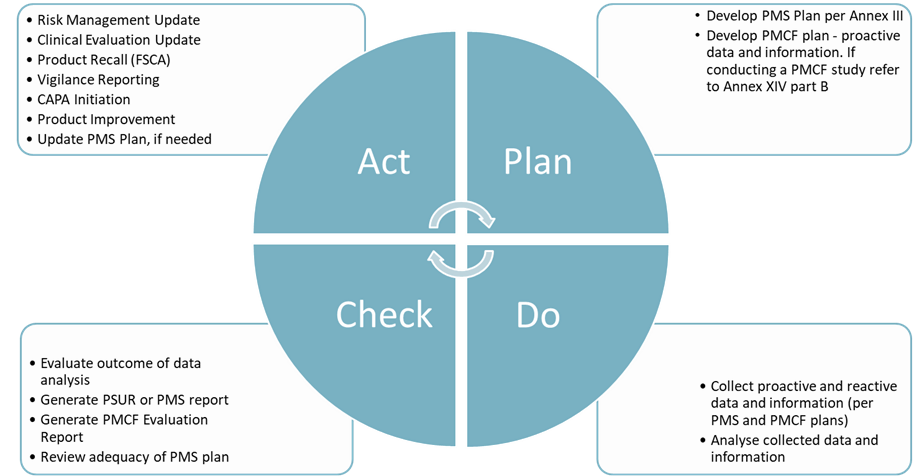
Figure 2: Application of the PDCA model to the management of a MDR compliant PMS system
PMS Process Interaction
You might have noticed that the PMS system does not operate in isolation – to operate effectively, the PMS system must interact with several QMS processes including but not limited to complaint handling, vigilance, risk management, clinical evaluation, design and development (improvement), regulatory reporting including EUDAMED (Refer to MDCG 2021-21 (Medical Device Coordination Group, 2021)) etc. Efficiency is key – delays or failures in data and information transfer, or issues locating supporting data and records, can lead to underreporting, overreporting, negative impact on patient safety, failure/delay in initiating a FSCA or CAPA if required and most importantly lack of regulatory compliance!
When setting up the PMS system, you need to consider how information will flow between the different QMS processes. It is useful to map out the process sequence and interactions, inputs and outputs using a tool such as SIPOC or any process mapping methodology. Also, a large amount of documentation will be generated from the PMS system and you may need to consider how/where these documents will be stored in accordance with the MDR retention requirements.
Don’t forget that the medical device QMS standard ISO 13485:2016 and risk management standard, ISO 14971:2019 also include requirements related to a PMS system.
Concluding Remarks
MDR introduces more prescriptive PMS requirements compared to its predecessor. The expectation is for manufacturers to adopt a total lifecycle approach for demonstrating device safety and performance. Reliance on complaints as the primary source of PMS data will not be acceptable under MDR – manufacturers need to carefully consider the requirements and plan how they will demonstrate compliance to the MDR and also how they will resource and maintain the system going forward.
Need support implementing an effective MDR compliant PMS process?
Trinzo is your one-stop-shop for global regulatory and quality compliance. Our industry-leading experts are on hand to support you in the creation of an MDR compliant PMS process and beyond. Book your free, no obligation consultation today.
References
- European Commission. (2013). Guidelines on a medical device vigilance system.
- Kearney, B., & McDermott, O. (2023). The Challenges for Manufacturers of the Increased Clinical Evaluation in the European Medical Device Regulations: A Quantitative Study. Therapeutic Innovation & Regulatory Science.
- Medical Device Coordination Group. (2020). MDCG 2020-7 Guidance on PMCF plan template.
- Medical Device Coordination Group. (2020). MDCG 2020-8 Guidance on PMCF evaluation report template.
- Medical Device Coordination Group. (2021). MDCG 2021-1 Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional.
- Medical Device Coordination Group. (2022). MDCG 2022-21 Guidance on Periodic Safety Update Report (PSUR) according to Regulation (EU) 2017/745.